

The study of the microscopic structure of animal and plant tissues, polymers and metals, requires sectioning of samples into extra-thin slices, which are then mounted into a glass slide and observed by microscopy. For most common applications, biological samples are dehydrated, fixed, and embedded into a support material such as paraffin or resin blocks. The block is then cut in an (ultra)microtome, yielding thin sections for microscopic observation. However, for some applications, it is not advisable to process the sample for paraffin/resin embedding, due to the generation of image artifacts and molecular degeneration that may occur due to the chemical treatment.
While paraffin/resin sectioning is better at retaining morphological features, cryopreservation and cryosectioning are indicated when the goal is to protect antigen and enzymatic integrity. In those cases, samples can be cryopreserved and cryosectioned, which maintains tissue/sample ultrastructure and does not require fixative reagents that may mask sample antigens. Thus, cryopreservation and cryosectioning are best suited for applications that involve the immunolabeling of samples, such as immuno-histo/-cytochemistry (IHC and ICC, respectively) and in situ probe hybridization (Fischer et al., 2008).
In this article, you will learn about different cryosectioning protocols, their applications and how to overcome common problems that arise during sample processing.
Sample freezing is achieved by immersing the sample in a bath of isopentane, then, storing it at -80ºC in a liquid nitrogen chamber. It is crucial that sample freezing occurs as fast as possible. Ultra-fast freezing ensures that water in the sample does not have time to form crystals, which are a common source of image artifacts. Thus, when samples are appropriately frozen, they become vitrified, and the final microscopic image is a faithful representation of the sample microstructure
Prior to cutting, samples are embedded in an inert material that serves as a support to place the sample in the cryotome. Sample holders have specific designs, thus, any tissue or tumor biopsy, or even small plant parts, must be embedded into a melted support medium (freezing matrix) in a mold (Fischer et al., 2008; Watkins, 2009). After solidification, the freezing matrix forms a block that fits the sample holder. Freezing matrices are generally made from polyvinyl alcohol, polyethylene glycol, and dissolved in a solution of other non-reactive ingredients (Wikipedia, 2016). They are sold under the general name OCT – Optimal Cutting Temperature Cutting compound, however, the amount of OCT must be tightly controlled, as too much OCT medium may lead to freezing artifacts.
This is the specific microtome used for cryosectioning. Cryotomes have roughly the same mechanical elements as other microtomes, plus they are supplied with cryostats, refrigeration systems that avoid sample melting. The blade and the surface where the thin sections are recovered are also maintained at cold temperatures, to avoid OCT melting before mounting the sample on the glass slide. Some cryotomes include thermal blocks that are kept at freezing temperature, where the mold with the OCT matrix plus the sample can be frozen, and where coated glass slides can be chilled, prior to receiving the cryosection (Figure 2).
Figure 2: The cryotome and its main elements. A. general view of the cryotome. B. Main elements inside the freezing chamber (image credit: Amos scientific).
After cut, cryosections are mounted into coated glass slides, where they can be further processed for staining, immunolabeling, in situ probe hybridization, etc. To promote better adherence of the cryosection to the slides, they are coated with positively charged cationic polymers such as poly-L-lysine, which help biological samples to adhere by attracting negatively charged molecules in cell surfaces.
At the beginning of a cryosection procedure, the first thing to set up is the cryotome itself. This step involves starting the device and setting up the temperature, which depends on the type of sample. Normal cutting temperatures are between −30ºC and –20ºC. Usually, softer tissues require colder cutting temperatures. Softer samples (e.g., brain tissue) are cut at –30◦C, while harder samples with a defined cytoskeleton (e.g., muscle) are cut at about –20◦C. For very hard specimens, it may be necessary to raise the temperature to –15◦C, although temperatures any warmer than this may lead to excessively soft specimen blocks (Watkins, 2009). Accordingly, both the blade and the coated slides, which will receive the cryosections, must be kept at low temperature, to avoid OCT melting during cutting and mounting, respectively.
The next parameter to set on the cryotome is the thickness of the sectioning. Initially, this is set to cut thicker sections (around 200 nm), for trimming the block until the blade reaches the sample itself. Then, the thickness must be adjusted. The thickness of the cryosections depends mostly on the downstream applications. In that context, if the sections are to be used for morphological studies and observed by optical microscopy, they must be cut at around 500 nm. Ultrastructural studies, such as transmission electron microscopy (TEM), require thinner sections to allow a beam of electrons across the sample, which usually varies between 30 nm and 100 nm (Miranda et al., 2015; Wikipedia, 2020).
Finally, before starting the cryosectioning, the operator must ensure that all additional elements are at hand proximity. These include a pencil (for sample identification in the glass slide), forceps (to remove the block with the sample and put it in the sample holder), and brushes (to remove debris from the cutting or to collect the cryosections and mount them in the coated slides).
Tissue/sample preparation for cryosectioning is a key step to obtain quality images. Cryosectioning can be used to cut samples fixed with cross-linking agents (such as 4% paraformaldehyde), and cryopreserved samples, in which fixation is done after sectioning. The type of sample fixation depends on subsequent applications. For some applications, tissue morphology is better preserved if the tissue is fixed and immersed in a sucrose solution. For that, the sample should be incubated in room-temperature fixative solution (usually 2% formaldehyde). The incubation time depends on the sample size and density. After fixation, the sample must be washed in phosphate-buffered saline solution (PBS), and immersed into a 0.5 M sucrose solution until it sinks (which usually takes between 3 h to 24 h). As cryosectioning is more often used to cut cryopreserved samples, we will describe the cryopreservation protocol in more detail.
Once the sample is cryopreserved in the OCT block, it must be mounted into the sample holder, after which it is ready for processing. Here, we describe the general steps of a cryosection protocol (Fischer et al., 2008; Watkins, 2009);
| Problem | Possible cause | Solution |
| Poor morphological
preservation throughout tissue |
Inadequate care during dissection or excessive delay between dissection and freezing | Take great care during dissection not to damage tissue by stretching or excessive bending. Freeze tissue soon after dissection to prevent proteolysis by endogenous proteolytic enzymes. |
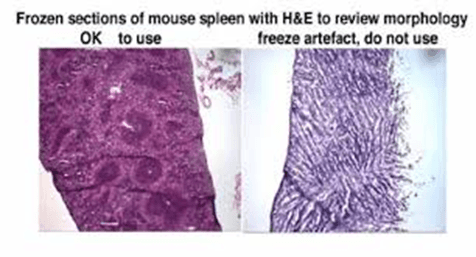
| Holes in tissue | Ice crystal formation in tissue during freezing, or due to thawing of the block and subsequent refreezing and ice crystal formation in the cryostat | Ensure that blocks are frozen in isopentane rather than a gas such as nitrogen. The latter will boil on contact with the warm tissue giving a “shell freezing” effect, where the boiling gas insulates the outer layers of the tissue from the cold liquid nitrogen, thereby slowing freezing and allowing ice crystal formation. Ensure that when specimens are mounted in the microtome, they are protected from warming by a large pre-cooled heat sink. Ensure that you do not touch the specimens with your fingers or with warm tools. |
| Morphology appears fuzzy | “Pressure artifact” due to
excessive pressure on the back of the slide while picking up the section |
Use less pressure when picking up the section. A mixture of warmth and a static charge will normally ensure that sections stick to the slide. If this is not the case, the slide is too cold and may be locally warmed by touching a finger to the back of the slide. |
| Morphology appear smeared | The slide was moved while lifting section | Hold the slide against the rear of the knife and use this as a fulcrum while gently rocking the slide down to touch the section, thereby ensuring no lateral movement of the slide. |
| Sections do not stick to slide; they wash away during labeling | Treated slides were not used | Use coated slides |
| Sections blow away from knife-edge during sectioning | A buildup of static electricity in the cryostat | There is no reliable solution. It is possible to buy polonium brushes, which may help. Antistatic guns (such as those designed for record players) have also been used successfully. |
| Sections have lines running up them and separate into ribbons on the knife-edge | Knife is dull | Replace knife, generally disposable knives are used routinely nowadays |
Thin-sectioning of biological samples, followed by chemical fixation to preserve ultrastructure features for microscopic observation, is a routine laboratory procedure. However, the process might pose some limitations to downstream applications. For example, the generation of artifacts due to poor cutting, or the masking of antigens by alcoholic and cross-linking reagents are commonly encountered problems. While the first might be resolved by appropriate microtome and blade maintenance, antigen masking depends on the sample ultrastructure and the reagents (antibodies) used. As such, avoiding antigen masking sometimes involves laborious and time-consuming optimization.
Cryopreservation, and cryosectioning, are straightforward techniques used to overcome the above limitations posed by more traditional sectioning and fixation protocols. The cryotome is a specific microtome, equipped with a refrigeration system, and cold blocks that keep the sample between -10ºC and -20ºC during sectioning. Importantly, the cutting blades are maintained at below zero temperatures, to avoid sample heating during cutting. Despite being a relatively straightforward technique, cryosectioning, if not performed appropriately, might also damage samples. Moreover, inappropriate handling of the cryotome, or poor maintenance of the cutting blades, often result in damage to the device, thus requiring expensive repair by specialized technicians. Therefore, the operation of the cryotome and the manipulation of the cryosections must be done by an experienced user, to obtain clean-cut sections that will provide artifact-free images.


